我们的基因体时代-AI, Data和生物资讯 Day15- 组装後的序列档案格式SAM, BAM
上一篇我们的基因体时代-AI, Data和生物资讯 Day14- 第二代定序(次世代定序)和它的资料格式上一篇介绍次世代定序之後的档案格式fastq和相关的软件和分析工具,接者再往下来到越来越复杂的序列拼装後的档案格式:bam和sam,这可能也要先介绍一下背景知识。
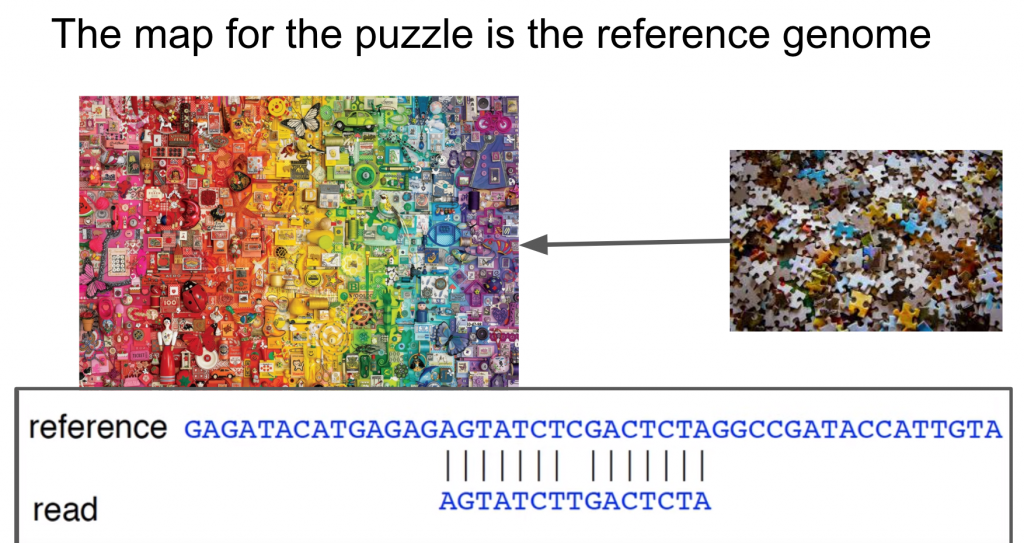
从上一篇我们知道从第二代定序机器出来的档案fastq中,可能会存放上百万个序列,是从定序机器得到的资料,但这些片段是来自於哪一个染色体或是位置,是不知道的,必须用演算法去做对照,有点像是拼拼图一样,如同下图:
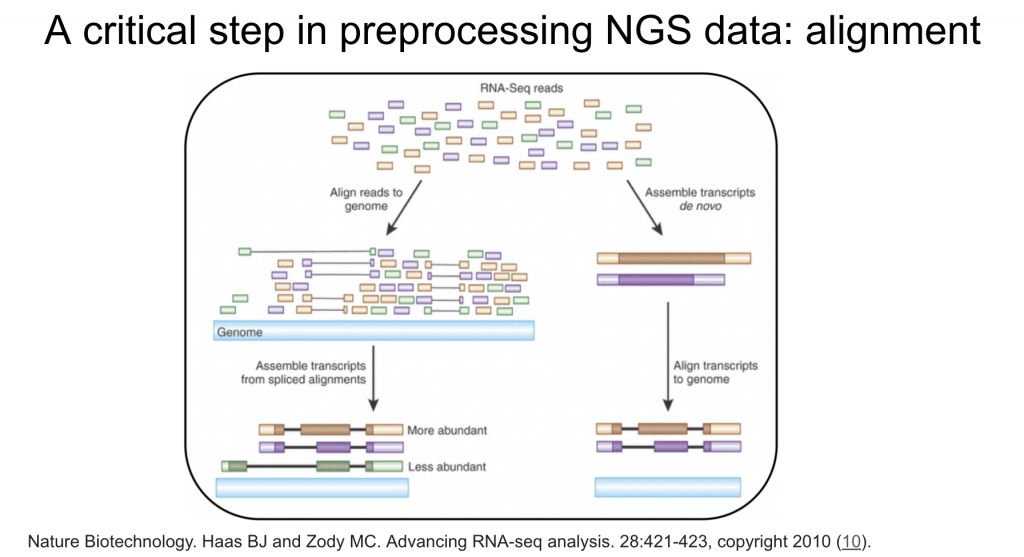
所以辨认每一个定序出来的片段来自何方的时候,我们必须要有个参照序列,就是所谓的参考基因组,但这个参考基因组跟实际上的定序对象的序列是有差异的(人跟人都会有基因序列的差异,所以用的参考基因组也会跟片段有所差异,所以这个比对是有一定的匡松条件,才能找到那些个人的变异资讯)。
所以当我们把这些片段比对到参考基因组上时,就必须要有另一个格式来储存这样的资料。
Sequence Alignment Map(SAM)序列比对格式
SAM这个格式就是为了要把比对参考序列的资料储存起来,而BAM档就是其二进位的格式,因为假如所有资料都是以text档案格式存放的话,他实际的档案大小是非常大的。提到SAM档案格式就要提到一个领域大神Heng Lin,他撰写的演算法和软件基本上是目前所有第二代定序分析软件的基石,像是bwa, samtools, 在2009年这篇论文中,Heng Lin提出的SAM档案格式也算是解决领域内资料统合的问题(有点像是fastq或是fasta格式的提出,也是为了统一当时的生物资料交流格式)。
详细的SAM格式,可以看这份最新的spec文件。可以看SAM格式说明书中的简易范例
@HD VN:1.6 SO:coordinate
@SQ SN:ref LN:45
r001 99 ref 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG *
r002 0 ref 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *
r003 0 ref 9 30 5S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0;
r004 0 ref 16 30 6M14N5M * 0 0 ATAGCTTCAGC *
r003 2064 ref 29 17 6H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1;
r001 147 ref 37 30 9M = 7 -39 CAGCGGCAT * NM:i:1
简单来说,SAM是一个tabl分隔的文字资料格式,其分成两个部分:
- Header
- Alignment
Header的部分,每一行是一笔资讯,且是以@开头,里面有储存整个比对中参考的染色体,@後面会有两个文字作为header资讯种类,至少有@HD(基本文件说明), @SQ(参考序列), @RG(片段分组), @PG(所用的比对分析程序和所下参数), @CO(注解)。




反之,在Alignment的部分则是以tab相隔的格式,里面共有11栏,每一栏的内容如下:

第一栏QNAME:代表此片段的独立名称,第二栏FLAG:为独立编码来代表此片段的比对状况,详细数字对应其状况,可由下面的图表来理解
 ,第三栏RNAME:代表此片段比对到的参考基因组其名字为何,通常就是对应@SQ的名称,第四栏POS:代表所对应到参考基因组的起始位置(从1开始的索引系统1-based system),第五栏MAPQ:代表期比对品质,数字越小代表此片段比对错误的机率越大,第六栏CIGAR:代表此片段跟参考序列之间的关系为何,也是有一个表可以对照
,第三栏RNAME:代表此片段比对到的参考基因组其名字为何,通常就是对应@SQ的名称,第四栏POS:代表所对应到参考基因组的起始位置(从1开始的索引系统1-based system),第五栏MAPQ:代表期比对品质,数字越小代表此片段比对错误的机率越大,第六栏CIGAR:代表此片段跟参考序列之间的关系为何,也是有一个表可以对照
 ,第七栏RNEXT:代表这个片段的另一组其所比对到的参考基因组名称(这跟定序方法有关,有所谓的paired-reads,此时也是@SQ的名称为止),第八栏PNEXT:则是此片段的另一组片段其比对到的位置(1-based system),第九栏TLEN:所在正股还是负股,第十栏SEQ:序列本身,第十一栏QUAL:品质资料。
,第七栏RNEXT:代表这个片段的另一组其所比对到的参考基因组名称(这跟定序方法有关,有所谓的paired-reads,此时也是@SQ的名称为止),第八栏PNEXT:则是此片段的另一组片段其比对到的位置(1-based system),第九栏TLEN:所在正股还是负股,第十栏SEQ:序列本身,第十一栏QUAL:品质资料。
理解这个资料格式的内容,那麽接下来就可以分享用来处理和分析相关资料的工具!
相关参考阅读:
我们的基因体时代:samtools 使用
我们的基因体时代:SAM, BAM and CRAM
Sequence Alignment/Map Format Specification
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-9. doi: 10.1093/bioinformatics/btp352. Epub 2009 Jun 8. PMID: 19505943; PMCID: PMC2723002.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H, Twelve years of SAMtools and BCFtools, GigaScience (2021) 10(2) giab008
这个月的规划贴在这篇文章中我们的基因体时代-AI, Data和生物资讯 Overview,也会持续调整!我们的基因体时代是我经营的部落格,如有对於生物资讯、检验医学、资料视觉化、R语言有兴趣的话,可以来交流交流!
<<: 15.移转Aras PLM大小事-Part料号全域搜寻
Day7 用python写UI-聊聊标签Label方法(二)
今天要主要会介绍几个 widget 的共通方法,後面的部分会介绍如何在介面上加上图片还有其他几个实用...
html 音量拉杆
今天来说要如何做出一个可以左右滑的拉杆,程序码范例如下 <input type="r...
[Day1] 简单介绍 Google Assistant 语音应用程序
大家好,我是Hank。 目前就读於台科大资工所的研究生。 很高兴有机会向大家分享我在开发Google...
第一天 参赛宣言
经历了前两次的失败,决定还是第一天不要直接写文章! 换个心情,先写了参赛宣言。好好的展开一个挑战的开...
实体关联性模型图 ER/EER Diagram
ER Diagram (Entity Relationship Model) 是一个非常热门的资料库...